
Warning: the blobulator is best viewed on a computer or tablet, not a smart phone.
Select input method:
Welcome to the Blobulator
Blobulation is an approach for detecting hydrophobic modularity in protein sequences based on contiguous hydrophobicity, originally developed by the Brannigan Lab for aiding in analyzing residue contacts in molecular dynamics (MD) simulations of a long intrinsically disordered protein (the BDNF prodomain). The blobulator allows the user to blobulate any sequence, and visualize the results while adjusting the two tunable parameters to detect blobs found at varying resolutions.
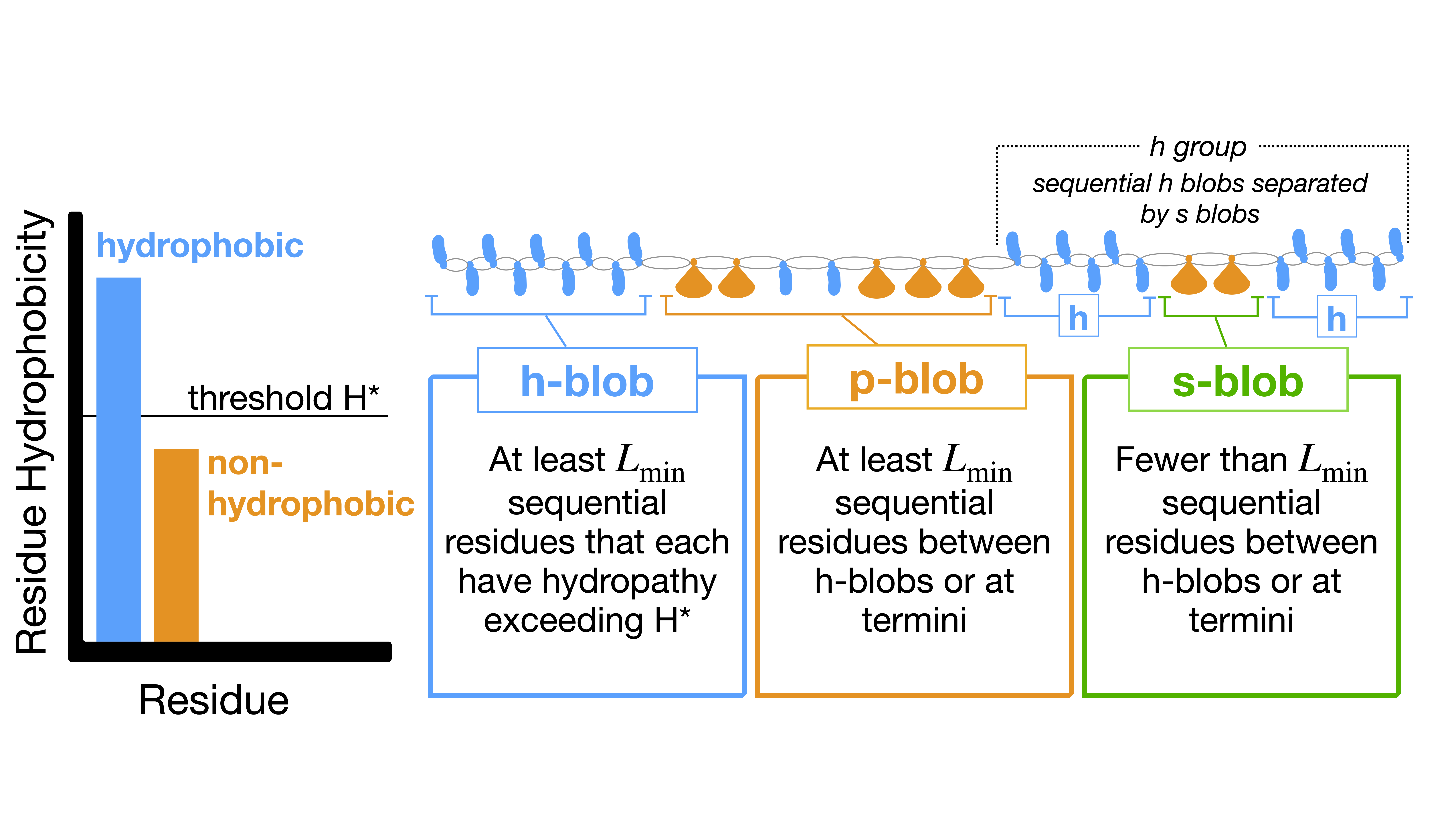
The figure above shows an overview of the blobulation algorithm. It consists of two steps - digitization and clustering. In the digitization step, residues are classified as either hydrophobic (blue) or non-hydrophobic (orange) by comparing their hydropathy to the user-selected threshold, H*. Next, the sequence is segmented: all segments containing only hydrophobic residues longer than Lmin are h-blobs (blue), other segments longer than Lmin are p-blobs (orange), and segments shorter than Lmin and non-hydrophobic are s-blobs (green). Figure adapted from Lohia et al. 2022.
In addition to outputting a visualization of the blobulated sequence, the blobulator also outputs several tracks showing blobs overlaid with additional information including: net charge, globular tendency (Das-Pappu phase), distance from the Uversky boundary, and sensitivity to mutation. These properties are also dynamically adjusted as the user increases or decreases resolution of the sequence. For human proteins, users will also see the location of known disease-associated single nucleotide polymorphisms (SNPs).
For bug reports, feature requests, or anything else - please contact at connor.pitman@rutgers.edu or grace.brannigan@rutgers.edu, or post an issue on our GitHub.
Citing the Blobulator
A manuscript on the Blobulator is currently under review. For now, please cite: Connor Pitman, Ezry Santiago-McRae, Ruchi Lohia, Kaitlin Bassi, Thomas T. Joseph, Matthew E.B. Hansen, Grace Brannigan, “The blobulator: a webtool for identification and visual exploration of hydrophobic modularity in protein sequences.” bioRxiv. 2024.
Sequence Input
The first thing needed to use the blobulator is either a UniProt ID for a protein of interest, or a manual sequence entry. UniProt IDs are our recommended format for retrieving the sequence required for blobulation. Above each “compute button” there is a manual text entry box, in which the you insert either your sequence or ID. Then press the “Compute” button.
Visualization
On the blobulation output page, there are three adjustable parameters:
The first step of blobulation is digitization. The algorithm first calculates each residue's hydrophobicity on the selected scale and then smooths this over the hydrophobicity of the adjacent residues. Residues are then digitized to hydrophobic or non-hydrophobic based on their smoothed hydropathy's relation to the hydropathy cutoff (shown as grey bars in the first track). This cutoff (shown as a blue line in the first track) can be adjusted by manual entry into the text box to the left, by adjusting the slider to the desired value, or by clicking a residue letter (found above the slider) to set the cutoff to a given amino acid's hydrophobicity on the selected scale.
This setting establishes a threshold for how many residues constitute blob. If a group of contiguous residues over the minimum blob size individually have smoothed hydrophobicities above the value for hydrophobicity cutoff, the cluster will be considered a ‘h’ (hydrophobic) blob. If not, a group will be considered a ‘p’ (non-hydrophobic) blob if the length is above the minimum blob size, or an 's' (short) blob if the length is below the minimum blob size. The hydropathy cutoff can be adjusted by manual entry into the text box to the left, or by adjusting the slider to the desired value.
This option is used to change a residue within the sequence, and see what potential effects it would have on the blobulation output. To mutate a residue, select which residue you would like to mutate, choose the amino acid you would like to mutate the residue into, and click the checkbox. Alternatively, the user can select one of the known disease associated single nucleotide polymorphism by clicking on a black triangle found at the bottom of each track. These will only appear if a UniProt ID is used.
SNPs and Mutations
Known disease-associated mutations are shown by the black triangles. Hovering over these mutations will display the amino acid change caused by them, as well as a clickable reference SNP cluster ID.
Interpreting Plots
After blobulation, multiple visualizations are produced.
This plot shows the smoothed hydropathy per residue. The core of blobulation consists of two parameters - the first being a hydropathy threshold. This threshold is shown by the blue line on the “mean hydropathy” axis. This line shows the hydropathy threshold (shown by a blue line), which determines the boundaries of the h and p blobs. Note that this graph is the only one that shows the residues individually, and can be used as a reference to how the residues are grouped together based upon their position above or below the blue line. If a group of contiguous residues over the minimum blob size individually have smoothed hydrophobicities above the value for hydrophobicity cutoff, the cluster will be considered a ‘h’ (hydrophobic) blob. If not, a group will be considered a ‘p’ (non-hydrophobic) blob if the length is above the minimum blob size, or an 's' (short) blob if the length is below the minimum blob size.
This second outputted visualization shows the blobs colored according to their globular tendency based on their Das-Pappu classification. The Das-Pappu phase diagram was originally used to estimate how a disordered sequence might behave based on the charge content. Each blob is colored according to the region they fall in Das-Pappu phase diagram. Specifically, these are: globular, Janus/boundary, strong polyelectrolyte, strong polyanion, and strong polycation. The height of each bar corresponds to their identity of either a "p" "h" or "s" blob.
This third outputted visualization shows the blobs according to their residues’ collective average charge. Each blob is evaluated based on its fraction of both positively and negatively charged residues. The darker blue a blob is shown here, the higher the fraction of positively charged residues are present within the blob. Alternatively, the darker red a blob is shown here, the higher the fraction of negatively charged residues are present within the blob. An even fraction of positive or negatively charged residues, or a low fraction of any charged residues results in a grey color.
This fourth outputted visualization shows the blobs according to their positions on the Uversky diagram , where the line between ordered and disordered is plotted. Calculated negative values (represented in orange) are more ordered and positive values (shown in blue) are more disordered.
This fifth outputted visualization shows the blobs according to their enrichment in documented disease associated SNPs (dSNP). This idea was investigated in the context of aggregating and non-aggregating proteins at various blob lengths and hydrophobicity cutoffs in our recent publication, from which the figure below is presented (Lohia, et al 2022).
This sixth and final outputted visualization shows the blobs according to their predicted fraction of disordered residues, which utilizes the Database of Disordered protein prediction . This disorder calculation is only available if the user uses the UniProt ID.
Saving Data
After the “Download data!” button (located just below the three adjustable parameters) is pressed, the raw data used to generate the tracks will be downloaded in the form of a csv file. Each column corresponds to one of the following: residue name, residue number, window, hydropathy cutoff, minimum blob size, average hydrophobicity, blob type, blob index number, blob Das-Pappu classification, blob net charge per residue, fraction of positively charged residues, fraction of negatively charged residues, fraction of charged residues, Uversky diagram score, blob dSNP enrichment, and blob disorder score.
Frequently Asked Questions:
The nomenclature used here comes from polymer physics (Pincus, 1976; de Gennes, 1979): a blob is a group of sequential monomers in a polymer chain that "clump" with a characteristic length. For more information see Scaling Concepts in Polymer Physics by Pierre-Gilles de Gennes.
Blobs here are determined by defining clusters of contiguously hydrophobic residues, and the non-hydrophobic residues that span between them. A blob is a contiguous stretch of either hydrophobic or non-hydrophobic residues greater than (or below) a certain length.
While many analyses exist that consider charge, disorder, or conformational states of proteins, the blobulator considers hydrophobicity and its role in the determination of regions of a protein. This has been shown already to be a powerful tool for analysis of different domains of the BDNF protein , the research within which this tool was developed. Additionally, our recent publication has demonstrated other contexts in which the blobulator has proven useful, particularly with regard to disease-associated SNPs.
The blobulator outputs 6 tracks showing blobs overlaid with additional information. Each graph shows the sequence of the protein displayed in one of the following ways: smoothed hydropathy per residue, colored according to globular tendency, colored according to net charge per residue, colored according to the Uversky diagram, colored according to dSNP enrichment, colored according to fraction of disordered residue.
We strongly recommend using the UniProt ID option when available. There will be more graphs outputted, as well as SNP data available if you blobulate the protein using its UniProt ID. If you are interested in a specific variant of the protein, such as one containing a SNP, there is a mutate residue option at the top of the output page.
For smaller proteins, we expect blobulation to take seconds. For very large proteins, the blobulator may take over a minute to produce a result.
To zoom in, hover your mouse over the plot you're interested in and click and drag around the area you wish to zoom in on. To revert the plot back to it's original zoomed-out state, double click on the plot.
These letters represent amino acids. Clicking a letter will set the hydropathy cutoff to the hydrophobicity of the selected amino acid for the chosen scale.
Yes! We recommend saving the page as a .pdf file using the “print” function in your browser.
The data can be downloaded using the “Download data!” button at the top of the blobulator output page. The downloaded data will be in the form of the csv file with labeled columns, which can be used to generate custom graphs or retrieve specific values.
Yes. Any adjustments made after blobulation but before the “Download data!” button is clicked will be reflected in the csv file.
It is possible that you chose the manual sequence entry option, for which there will be no SNP data, or that there is no data in EMBL-EBI for your protein of interest. It is also possible that you are not blobulating a human protein. In any of these cases, it doesn’t necessarily mean that no SNP data exists for the protein you are blobulating.
Check and make sure you chose the UniProt ID input option.
Please contact us and let us know what you’re thinking. Our goal is to maximize the blobulator’s usefulness, and any suggestions are greatly appreciated. In the meantime, the local version of the blobulator, which can be found on our github, can be modified to your liking.
Yes! It can be found on our github.
We recommend using our back end code found on our GitHub. If you think a specific feature would be generally useful, please email us at connor.pitman@rutgers.edu or grace.brannigan@rutgers.edu.